































Dr. Theodore Roth
Marson研究室(University of California San Francisco)
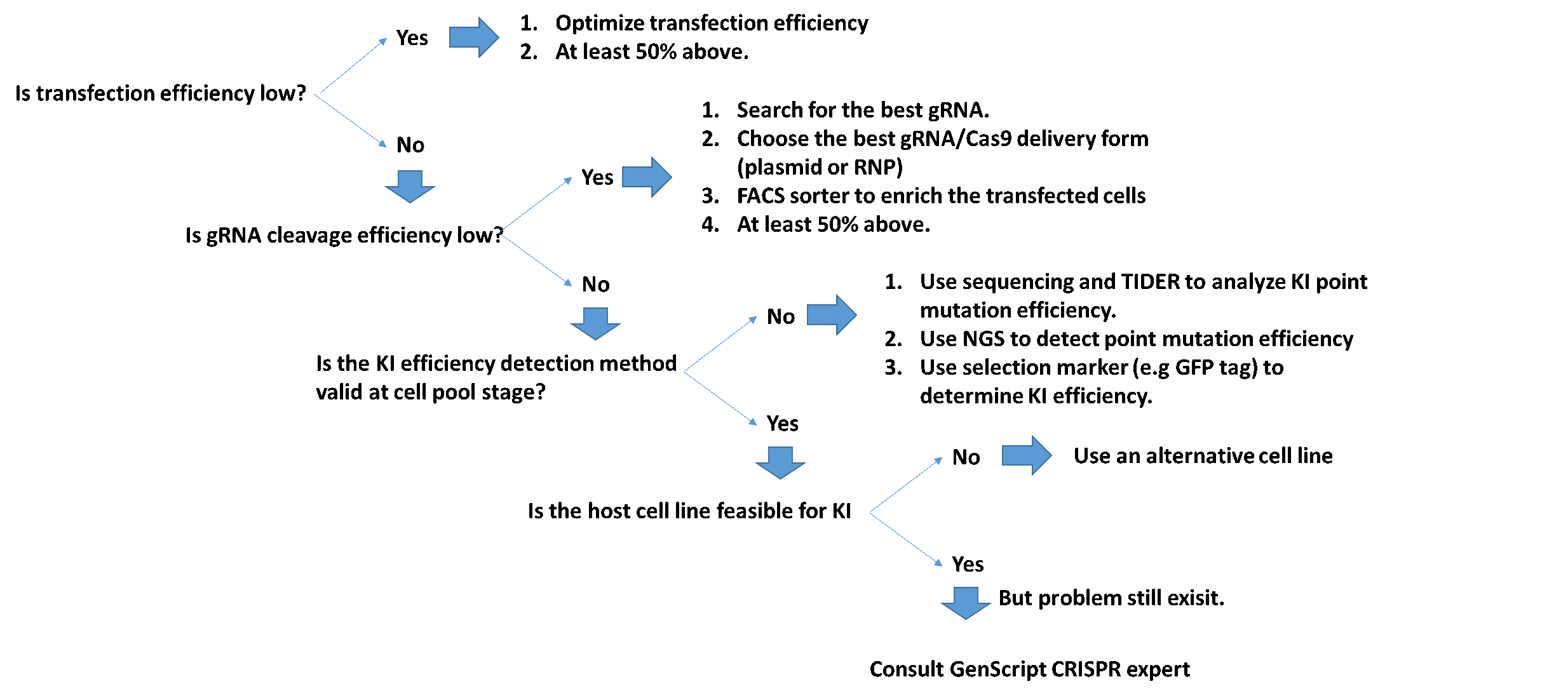
遺伝子編集実験で特に必要とされる高濃度の長鎖ssDNA配列は、研究室での合成が難しいのですが、GenScriptの長鎖ssDNAを使用して、ヒトプライマリーT細胞への長鎖DNA配列導入に成功できました。

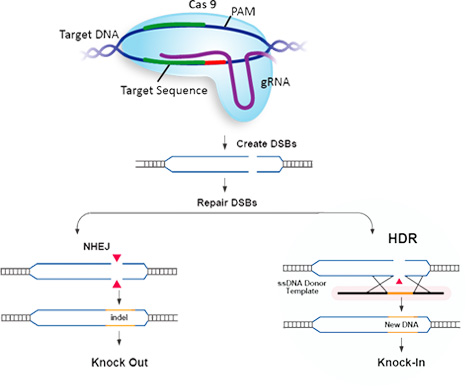
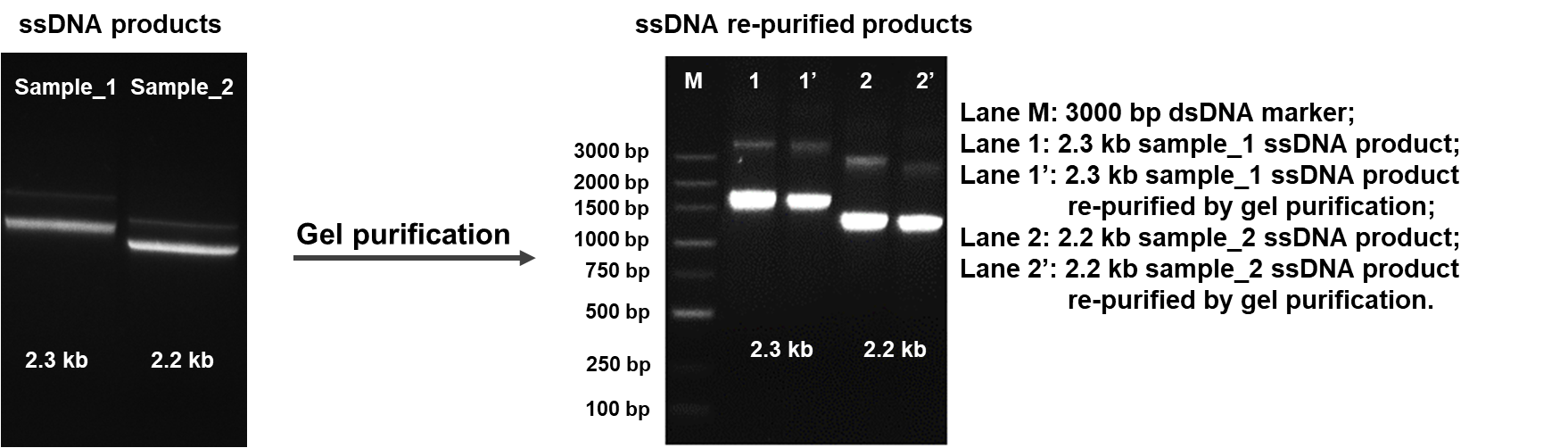
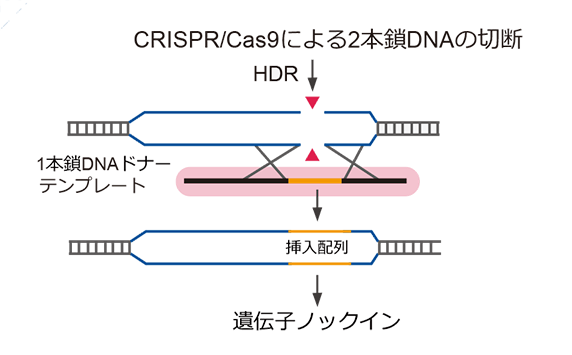
一本鎖DNA(ssDNAまたはssODN)は、高い編集効率かつオフターゲットを減少した、遺伝子ノックイン作製のための最良なCRISPR相同組換え修復(HDR)テンプレートとして、皆様にお使いいただいています。GenScriptは、お客様のCRISPR実験の編集効率を最大化するために、高品質で配列検証済のssDNAを提供しています。

Dr. Theodore Roth
Marson研究室(University of California San Francisco)
遺伝子編集実験で特に必要とされる高濃度の長鎖ssDNA配列は、研究室での合成が難しいのですが、GenScriptの長鎖ssDNAを使用して、ヒトプライマリーT細胞への長鎖DNA配列導入に成功できました。
ssDNA
最速3週間でお届け
| Quantity (ug) | <500nt (JPY/Item) | 501-5000nt (JPY/Item) |
|---|---|---|
| 3 | ¥64,000 | ¥64,088~¥472,000 |
| 5 | ¥80,000 | ¥80,096~¥552,000 |
| 10 | ¥104,000 | ¥104,104~¥624,000 |
| 20 | ¥120,000 | ¥120,112~¥704,000 |
| 50 | ¥192,000 | ¥192,128~¥984,000 |
| 100 | ¥240,000 | ¥240,136~¥121,6000 |
| >100 | お問い合わせください | |
5 kbより長い配列は、 [email protected]までお問い合わせください。
*液体製品を納品する場合に、追加送料があります。
| 試験内容 | 試験方法 | 合格基準 | リサーチグレード (≤2mg) |
|---|---|---|---|
| 純度 | アガロースゲル電気泳動 | 単一バンド | ✔ |
| シークエンスの正確性 | サンガーシークエンス | 100%正確であること | ✔ |
| 吸光度 | 分光光度計 260 nm/230 nm | ≥ 2.0 | ✔ |
UCSF(University of California San Francisco)のMarson研究室との共同研究で、GenExact™ ssDNAは研究室内で生成したHDRTs(相同組換え修復テンプレート)を一貫して上回り、より低レベルの毒性と高いノックイン効率を示しました。エレクトロポレーションによる、エンハンサーを使用しないGenExact™ ssDNAのノックイン効率は、GMP互換スケールで46.2%に達しました。GenExact™ ssDNAで生成されたノックイン陽性細胞の数は、想定患者投与量(100*10e6)をはるかに上回りました。In vitroアッセイではBCMA-CAR細胞による効率的な細胞死が証明されました。
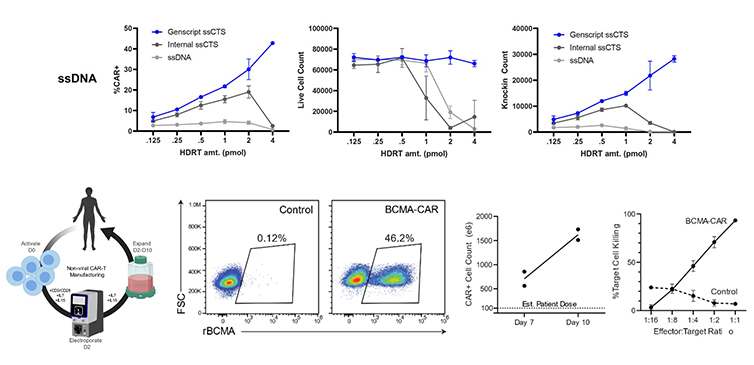
Brian R Shy. et.al., bioRxiv(2021), Read the whole article.
![]() CRISPR Knock-in Protocol for HEK 293T/293 cells with Thermo Fisher Neon®
Transfection System
CRISPR Knock-in Protocol for HEK 293T/293 cells with Thermo Fisher Neon®
Transfection System
![]() CRISPR Knock-in Protocol for Jurkat cells with Thermo Fisher Neon® Transfection
System
CRISPR Knock-in Protocol for Jurkat cells with Thermo Fisher Neon® Transfection
System
![]() CRISPR Knock-in Protocol for stimulated Human T Cells with Lonza 4D Nucleofector™
X Unit
CRISPR Knock-in Protocol for stimulated Human T Cells with Lonza 4D Nucleofector™
X Unit
![]() CRISPR Knock-in Protocol for stimulated Human T Cells with Maxcyte® Electroporation System
CRISPR Knock-in Protocol for stimulated Human T Cells with Maxcyte® Electroporation System